A large number of problems in ab‐initio quantum chemistry involve finding the global minimum of the total system energy. These problems are traditionally solved by numerical approaches equivalent to local optimization. While these approaches are relatively efficient, they do not provide guarantees of global optimality unless a starting point sufficiently close to the global minimum is known apriori. Due to the enormous amount of computational effort required to solve these problems, more mathematically rigorous alternatives have so far received very little attention. Taking the above issue into consideration, this paper explores the use of deterministic global optimization in the context of Hartree‐Fock theory, an important mathematical model applied in many quantum chemistry methods. In particular, it presents a general purpose approach for generating linear relaxations for problems arising from Hartree‐Fock theory. This was then implemented as an extension to the
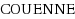 (Convex Over and Under ENvelopes for Nonlinear Estimation) branch and bound mixed integer non‐linear programs solver. Proof of concept calculations that simultaneously optimise the orbital coefficients and the location of the nuclei in closed‐shell Hartree‐Fock calculations are presented and discussed.
(Convex Over and Under ENvelopes for Nonlinear Estimation) branch and bound mixed integer non‐linear programs solver. Proof of concept calculations that simultaneously optimise the orbital coefficients and the location of the nuclei in closed‐shell Hartree‐Fock calculations are presented and discussed.
